Karbapenemek 2.
Kémiai tulajdonságok
Összehasonlítva a penicillinekkel vagy kefalosporinokkal, a karbapenemeknek jóval kevesebb utólagos szubsztitúciós reakcióját és egyéb manipulációjátt írták le. Cserébe viszont rendkívüli energiát fektettek a különböző cégek a totálszintézisbe. Ennek oka, hogy igen érzékeny vegyületek lévén, a legtöbb változtatást a totálszintézis korai fázisában, az indolin gyűrű zárása előtt kell elvégezni. A feszült β-laktám gyűrű miatt a vegyületek protikus kőzegben igen érzékenyek, például az asparenomicin enyhe savas kürülmények között néhány óra alatt elbomlik:

A szabad aminocsoportot tartalmazó származékok, pl. maga a tienamicin, vizes oldatban semleges körülmények között is igen gyorsan elbomlanak (gyorsabban, mint azonos körülmények között a G-penicillin), különösen magasabb koncentráción, ennek oka az amino csoport intramolekuláris támadása a β-laktám gyűrű ellen.
A C-1 atom szubsztituálása teljes szintézissel oldható meg. A bioaktivitást is figyelembe véve, gyakorlatilag csak az 1β-metil származékok érdekesek. Aktívak még az 1-fluoro vagy 1,1-difluoro származékok is, ezek azonban kémiailag nem elég stabilisak. A C-2 szénatom szubsztituense ugyanazt a szerepet játssza, mint a penicillinek és kefalosporinok 6(7)-acilamido oldallánca, vagyis a hatás spektruma és erőssége széles határok között módosítható ennek változtatásával. A természetes származékokban, mint láttuk, ciszteamin van ezen a helyen, illetve ennek oxidált, telítetlen vagy acilezett változatai. Bár a természetes oldallánc eltávolítható hipobrómossavval és a képződő 2-SH származék újból szubsztituálható, a természetes alapanyag szűkössége miatt az igen nagyszámú módosított vegyületet gyakorlatilag a következő három módszerrel állítják elő;
b) A másik szintén gyakori eljárásnál a gyűrűzárás előtt már beviszik a -SR csoportot tioészter formájában, s ezután zárnak gyűrűt Wittig vagy Wittig-Horner reakcióval:
c) Újabban a 2-triflátok és sztannánok vagy boránok Pd° katalizálta kapcsolásával totálszintézis nélkül is előállíthatók a C-2 atomon telített vagy telítetlen szénláncot vagy gyűrűt tartalmazó analógok.
 Válassz:
Válassz:
A hármas helyzetben a karboxi csoport védésére csak olyan származékok jöhetnek szóba, amelyekből igen enyhe körülmények között regenerálható a sav. Gyakorlatilag kétféle észtert alkalmaznak: a 4-nitro-benzilt, amely enyhe katalitikus hidrogénezéssel bontható, vagy az allil-észtert, amely Pd° katalizálta hidrogéntranszferrel (Pd(PPh3)4, K-etilhexanoát) hidrolizálható.
Igen lényeges a C-6 hidroxi-etil oldallánc sztereospecifikus kialakítása, mivel a legtöbb karbapenem ezt tartalmazza. A természetes PS származékoknál csak etil csoport van e helyen, ezek is hatékonyak és ez lehetőséget adhat egy asszimetria centrum megspórlására a totálszintézisek során, azonban mint később kiderült, az etil származékok biológiai hatása elmarad annyival a hidroxietil mögött, hogy ez utóbbi megérje az előállítással járó többletmunkát. A hidroxi-etil csoport sztereospecifikus bevitelét általában a korai fázisban célszerű megtenni, kialakításának legkézenfekvőbb kialakítása acetaldehidnek a megfelelő anionnal való reakciójával biztosítható:

Ezt a módszert főleg a korai szintéziseknél alkalmazták, hátránya, hogy mivel két asszimetriacentrum is keletkezik egyidejűleg, a sztereospecifitás vagy csak királis reagensekkel biztosítható, vagy már eleve minél több királis centrumnak kell lennie a molekulában, azonban a reakció sztereokémiai lefutása így is nagyon függ a gyűrű szubsztitúciós viszonyaitól. Célszerűbb ezért a karbapenemek szintézisének kiindulási anyagaként penicillint vagy egyéb könnyen elérhető optikailag aktív vegyületet választani, lehetőleg olyat, ami sok analóg származéknak lehet a közös kindulási anyaga, s már ebben a korai fázisban megoldani a hidroxietil csoport kialakítását. Ezt a következő részben mutatjuk meg.
Totálszintézisek
Mint a következőkben is látni fogjuk, a totálszintézisek legtöbbjében az indolin gyűrű zárását az alábbi három mód valamelyikével oldják meg:
- Wittig reakcióval
- Diazoketonból kialakított karbénnel
- Dieckmann kondenzációval
A tienamicin első totálszintézisét a Merck kutatói publikálták 1980-ban. A szintézis L-glutaminsavból indul ki. Megfelelő védések után a β-laktámgyűrű kialakítását t-butil-magnézium-kloriddal végezték, majd a 4-oldalláncot maszkírozott savvá alakították. Ezután a β-laktám gyűrűre a jellemző hidroxi-etil oldalláncot acetaldehid addícióval lítiáláson keresztül alakították ki. Itt a reakciókörülmények megválasztásával lehetett biztosítani, hogy főleg a várt R-izomer képződjön. Ezután a 4-CH2COOH csoport kialakítása után a lánchosszabítást újszerű és azóta gyakran alkalmazott módon, a sav imidazolidjának egy megfelelő magnézium-malonát-észterrel történő reakciójával alakították ki. A kapott ketoésztert valamilyen arilszulfonilaziddal kezelve, a megfelelő diazo-származék képződik, amely ródiumkatalízissel egy karbénreakció során adja a gyűrűzárt terméket.
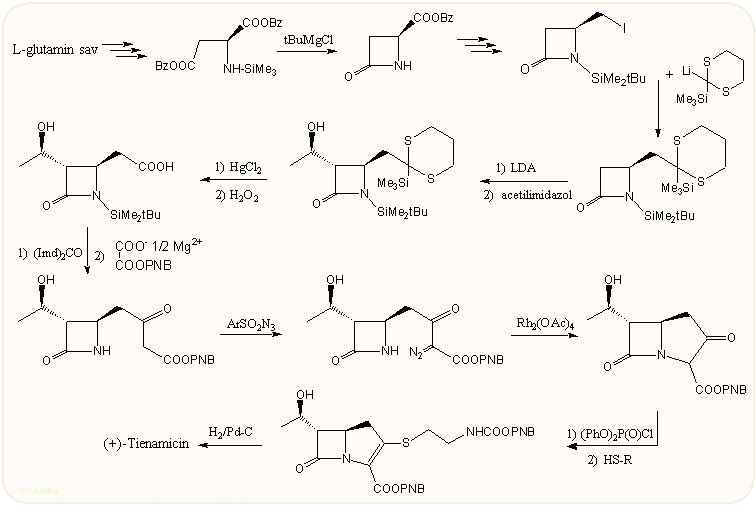
A biciklusos ketoésztert vinilfoszfát származékká alakítva és a megfelelő védett ciszteaminnal reagáltatva, majd a védőcsoportokat eltávolítva jutunk a végtermékhez. Ennek a Merck szintézisnek a mintájára igen sok variációt dolgoztak ki, amelyeknek főleg az első fele különbözik egymástól, vagyis a 4-CH2cOOH azetidinon kulcstermék kialakítása, míg a pirrolin gyűrű kettőskötésének és kettes helyzetű szubsztituensének kialakítása a már említett foszfát észteren keresztül az egyik legáltalánosabban használt eljárás.