Szerkezet-hatás összefüggések (QSAR) 2.
Free–Wilson-analízis
Előfordulhat olyan szituáció is, amikor nem állnak rendelkezésre megbízható extratermodinamikai paraméterek. A nem sokkal a Hansch modell után született FreeWilson modell alapfeltételezése az, hogy hatóanyagok egy homológ sorában a molekula egyes szegmensei additív és konstans módon járulnak hozzá a biológiai hatáshoz, másszóval az alapváznak van egy alap biológiai hatása (konstans szegmens), és a szubsztituensek ehhez additív és mindig azonos módon járulnak hozzá (változó szegmensek):
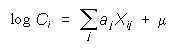
ahol C a mért biológiai hatás, aj a j-dik szubsztituens hozzájárulása C-hez, az Xij indikátorváltozó értéke pedig 0 vagy 1 aszerint, hogy egy bizonyos szubsztituens jelen van-e a j-dik helyen, μ az alapmolekula biológiai hatása. Az alábbi táblázat példájában fenil-etilamin származékok adrenerg receptor blokkoló hatását vizsgálták, a fenilcsoport szubsztitúciójának függvényében. Összesen 10 független változó van (x1 x10), a 22 egyenletből meghatározhatók a legjobb illeszkedés koefficiensei. Pozitív előjel esetén az adott szubsztituens növeli a hatást és fordítva.

Igy például a 14. egyenlet a következő:
A szubsztituensek hatáshozzájárulása nem teljesen független egymástól, azonkívül egyszubsztituensek. Ezeket további indikátorváltozókkal lehet figyelembevenni, pl. 1 ha van hidrogénhid a molekulában, 0 ha nincs.
Topológiai paraméterek, térszerkezet
Az előző módszerek bármelyikében problematikus a térszerkezet figyelembevétele. Egy n-butil és egy n-oktil csoport elektronikus hatása nem sokban különbözik, a hidrolízisadatokból származtatott ES értékek sem tükrözik a különbséget. Hasonlóan problémás például az alkilcsoportok izomeriájának a figyelembevétele, holott ez nyilván fontos egy receptorral való kapcsolódás esetén. Egy molekula térfogata skaláris mennyiség, a molekula alakja pedig már vektoriális. Az előbbit könnyen be lehet építeni egy regressziós egyenletbe, de az utóbbi matematikai kezelése nem egyszerű.
A térszerkezet figyelembevételének egyik módja az ún. Verloop-féle sterimol paraméterek használata. Ennél egy szubsztituenst hat változóval jellemeznek. Első lépésben megállapítják a csatlakozási pont (1) és a következő atom (2) irányában számítva a molekula(rész) legnagyobb kiterjedését (L), a van der Waals sugarak figyelembevételével. Ezután L-re merőlegesen, az y-z síkra vetítve megkeresik a legkisebb kiterjedést (B1), és az ábrán látható módon, B1 meghosszabításában ill. arra merőlegesen meghatározzák B2 - B4 -et. A legnagyobb L-re merőleges kiterjedés B5. L, B1 és B5 segítségével jó leírható a molekula eltérése a gömb alaktól:

Egy másik módszer a konnektivitási indexek használata, amit Kier vezetett be a QSAR gyakorlatába, s tulajdonképpen a gráfelmélet kémiai alkalmazásának egy módja. Ez azon a megfigyelésen alapszik, hogy egy molekula fizikai tulajdonságai különbözőképpen függnek a molekula konnektivitásától, topográfiájától. Például az izomer alkánok moláris refrakciója, vagy az alkoholok képződéshője alig függ a konnektivitástól, azonban az alkoholok forráspontja 20-30%-kal is változik. Az észterek vízoldékonysága egy nagyságrendet is változhat az izomeria, konnektivitás függvényében. A konnektivitást az ún. chi indexszel (χ) jelölik. Aszerint, hogy az egész molekulából, mint gráfból mekkora algráfot veszünk tekintetbe a számításnál (magyarán, hány kötésből számoljuk ki az indexeket), különböző rendű chi indexek léteznek (1χ, 2χ, stb.) Az alábbi ábrán látható, hogy miként kell kiszámolni a kötések ú.n. Randič-féle elágazási indexet, és ebből a molekula 1χ konnektivitási indexét:

Bonyolultabb molekulák esetén a kettőskötések duplán számítanak (fantomelágazás), heteroatomok esetén a magányos elektronok szintén fantomkötésként szerepelnek:

Magasabb rendű χ indexek számolásánál az összes lehetséges magasabb rendű algráfot tekintetbe kell venni, a trifluorecetsav példáján:

A konnektivitási indexek segítségével jó összefüggéseket lehetett találni vízoldékonyság, kromatográfiás retenciós állandók, forráshő, móltérfogat, stb. és a szerkezet között. Például különböző alkilszubsztituált fenolok mért és számolt toxicitása szintén jó egyezést mutatott, ha a Hansch-analízisben első és harmadrendű konnektivitásokat használtak:

Ez a tisztán topológiai index természetesen csak speciális esetekben használható jól, ezért Kier és Hall továbbfejlesztette olyan irányban, hogy az atomi kémiát és a kémiai környezetet is tartalmazza, tehát jelezze a p elektronok, magányos elektronok jelenlétét, stb., s megalkották az atomokat sokkal pontosabban jellemző ún. elektrotopológiai indexet, ennek taglalása azonban itt túl meszire vezetne.
Minta felismerés (pattern recognition, cluster analysis)

Ezt a statisztikai módszert – amelynek egyébként óriási az irodalma és a felhasználási területe – akkor lehet használni, ha a függő és független változók nem folyamatos értékeket szolgáltatnak, nagy a független változók száma, indikátorváltozókat használunk, úgyhogy ilyenkor inkább csoportosításról beszélhetünk. A pontos hatásmechanizmus általában nem ismert, és megpróbálunk valami kapcsolatot keresni a vegyülettel összefüggésbe hozható tulajdonságok között. A vegyületekhez ilyenkor nem folyamatos mérési adatok tartoznak, hanem n számú "molekuláris deskriptor", s azt keressük, hogy találunk-e ebben az n-dimenziós térben a függő változóval, például biológiai hatással egyértelműen korreláló halmazokat, osztályokat (clusters). Egy-egy vegyületet egy vektor jellemez, ahol n a deskriptorok száma:
A következő bemutatott példában 200 policiklikus aromás szénhidrogént (PAH) karcinogén hatását vizsgálták, pontosabban ezt a 200 molekulát használták tanítóként, majd az ezekből kapott összefüggéssel 400 véletlenszerűen kiválasztott PAH rákkeltő hatását vizsgálták. E vizsgálatban bináris döntést programoztak, vagyis a 200 tanító molekulát két csoportba osztották aszerint, hogy van vagy nincs rákokozó hatásuk:

Jurs, P. C., Chou, J. T. and Yuan, M.: "Studies of Chemical Structure-Biological Activity Relations Using Pattern Recognition" in: Computer Assisted Drug Design (Olson, E.C. and Christofferson, R. E. eds), chapter 4., ACS Symposium Series 112, Washington 1979 alapján
A molekulákhoz 28 szerkezettel kapcsolatos deskriptort rendeltek hozzá (egy részük látható a következő ábrán), ezek egy része a szerkezettel, alakkal kapcsolatos, mint a moltérfogat, konnektivitások, főtengelyek illetve ezek arányai, bizonyos szubstruktúrák (ábra) vagy heteroatomok jelenlétét vagy hiányát jelzi, vagy a megoszlási hányadost és a szubstruktúrák számolt s töltését stb. mutatja. A kapott modell prediktív képességét 400 molekulával vizsgálták, amelynek során a karcinogén molekulákat 87.5%, a nemkarcinogén molekulákat 92.5%-os pontossággal felismerte.


Ezek a típusú vizsgálatok már átvezetnek a mesterséges intelligencia témakörébe. Napjainkban egy ilyen módszer az öntanuló visszacsatolt neurális hálózatok (neural networks) alkalmazása a különböző fizikai, kémiai és biológiai hatások megbecslésére, ennek taglalása azonban túl messzire vezetne.
Összefoglalás
Az előzőekben tárgyalt módszerekről összefoglalóan a következőket mondhatjuk:
A regressziós egyenletekkel egy vegyületcsalád vagy homológ sor biológiai hatása többé-kevésbé jól leírható, általában egy szubsztituált alapmolekulából kiindulva. Látszik azonban, hogy ennél és a hasonló módszereknél több lényeges elhanyagolás, egyszerűsítés van, ami egyúttal korlátozó tényező a tervezés során:
- In vivo jónéhány tényező hat a gyógyszer sorsára: felszívódás, transzport, bejutás a sejtbe, eljutás a receptorhoz, kölcsönhatás a receporral. A klasszikus QSAR ezeket nem tudja igazából szétválasztani.
- Jobb az eset az in vitro vizsgálatok kiértékelésénél, hiszen itt már jóval kevesebb faktorral kell számolni. Jól használhatók a Hansch típusú egyenetek, ha pl. csak a szérumkötődést vizsgáljuk, vagy csak egy transzport folyamatot, vagy csak egy receptorral való in vitro kölcsönhatást.
- A statisztikai kiértékelés miatt minél nagyobb számú vegyület szükséges, és ismerni kell a szubsztituens paramétereket is.
- A módszer nem vesz figyelembe, nem tud figyelembe venni semmi "on-the-fly", vagyis menetközbeni, a receptorra való illeszkedéskor létrejövő konformáció változást.
- A konkrét molekuláris szintű gyógyszer-receptor kölcsönhatás itt nem ismert. Főleg emiatt a módszer csak egy vegyületcsaládon, homológ soron belül haszálható: másszóval, a módszerrel jól lehet már ismert "lead" vegyületeket optimalizálni, de nem lehet vele szerkezetileg új vegyületeket keresni.
Mindazonáltal e módszerekkel a hetvenes-nyolcvanas években igen nagyszámú analízist végeztek. A módszer alkalmazásával kapott regressziós egyenlet ill. annak grafikus ábrázolása megmutatja, hogy egy adott molekulasereg lipofilitási stb. tulajdonságait milyen irányba kell változtatni, hogy a hatást optimalizáljuk. A teljes biológiai hatás ("a" biológiai hatás) előreszámolásához, tervezéséhez ez a módszer tökéletlen, csak bizonyos feltételek együttes meglétekor lehetséges. Viszont kiválóan alkalmas egy-egy részlet tanulmányozására, becslésére, pl. a lipofilitási, ionizációs stb. viszonyoktól függő felszívódás, szérumkötődés stb. Röviden a módszer elsősorban egy vegyületcsaládon belüli optimalizálásra használható.