Kefalosporinok 3.
Félszintetikus kefalosporinok
A kefalosporinok és kefamicinek aminoadipil oldallánca enzimatikusan nem távolítható el, s arra sincs lehetőség, hogy a penicillinekhez hasonlóan, az oldallánc nélküli alapvegyületet (7-ACA) közvetlenül fermentációval kapják meg. A kémiai eltávolítás egyik első módszerénél nitrozilkloridot alkalmaznak, amikoris egy gyűrűs intermedieren keresztül képződik a szabad amin. A penicillineknél leírt iminoéteres módszer itt is alkalmazható, de az oldallánc eltérő jellege miatt többszörös védést kell használni, pl. a két karboxil csoportot benzhidril észterként, az amino csoportot acetil vagy ftalil származékként védik. Az iminklorid képzéshez oxalilkloridot vagy foszgént használnak. Érdekes japán szerzők megoldása, ahol oxalilkloriddal a kefamicin C-3-as aminocsoportját védik, a C-7-es amidot átacilezik, majd az oxalilamid csoportokat híg sósavas hidrolízissel ill. difenilkarbodiimiddel szabadítják fel. A kapott 7-ACMA közvetlenül felhasználható újraacilezéshez.

 Mivel a kefem alapvázhoz két ponton kapcsolódnak természetes szubsztituensek, a 7β-amino csoporton és a 3-metil csoporton, az alapváz két helyen is módosítható. Harmadik módosítási pont a 7α helyzet. Így az előállítható félszintetikus származékok száma is jóval nagyobb lehet a penicillinekhez viszonyítva, s a szerkezet-hatás összefüggések is bonyolultabbak. Nagyon általánosítva elmondható, hogy a 7-es amid szubsztituens inkább az antibiotikus hatásspektrumot befolyásolja, míg a 3-szubsztituens többnyire a farmakokinetikai viselkedésért felelős - ez azonban inkább csak tendencia, mivel erős az átfedés. Tény, hogy egy "rossz" 7β-oldalláncon nem nagyon lehet segíteni a 3-as helyzet változtatgatásával.
Mivel a kefem alapvázhoz két ponton kapcsolódnak természetes szubsztituensek, a 7β-amino csoporton és a 3-metil csoporton, az alapváz két helyen is módosítható. Harmadik módosítási pont a 7α helyzet. Így az előállítható félszintetikus származékok száma is jóval nagyobb lehet a penicillinekhez viszonyítva, s a szerkezet-hatás összefüggések is bonyolultabbak. Nagyon általánosítva elmondható, hogy a 7-es amid szubsztituens inkább az antibiotikus hatásspektrumot befolyásolja, míg a 3-szubsztituens többnyire a farmakokinetikai viselkedésért felelős - ez azonban inkább csak tendencia, mivel erős az átfedés. Tény, hogy egy "rossz" 7β-oldalláncon nem nagyon lehet segíteni a 3-as helyzet változtatgatásával.
Kémiailag nézve a gyógyászatilag is alkalmazott hatásos vegyületeket az alábbi összefoglalásokat tehetjük:
3-as helyzet: A természetes acetoxi vagy karbamoiloxi származékokat kevés vegyületben találjuk meg. Helyettük leggyakrabban merkapto-heteroaril csoportot vagy a negyedik generációs termékekben kvaterner ammónium vagy N-heteroarilium csoportot találunk. Érdekes, hogy a kvaterner nitrogént tartalmazó vegyületeket alkalmazták már a legkorábbi, első generációs származékokban is, majd a merkapto származékok megjelenésével eltüntek, végül a negyedik generációs extra széles spektrumú származékokban ismét előkerültek. Ezek a vegyületek csak parenterálisan alkalmazhatók. Az orális származékoknál a 3-as helyzetben hidrogén, metil, klór, metoxi vagy újabban vinil és propenil csoport van. Az orális hatás ára viszont a kisebb hatásspektrum. Ez úgy kerülhető ki, hogy egy jó parenterális származékból prodrogot készítenek aktív észter formájában. A kémiai változtatások in vivo hatását sokszor nehéz előre felmérni: például a cefalexin és a megfelelő 3-metoximetil származék in vitro hatása, valamint a per os felszívódási viszonyai gyakorlatilag azonosak. Ennek ellenére in vivo egérben kb 1/6-1/10 metoxi származék biztosítja ugyanazt a kemoterapeutikus hatást.
7α-helyzet: A hidrogén helyett metoxi (kefamicinek) vagy újabban formamido csoport lehet. E két helyettesítés a hatáserősséget valamelyest csökkenti, ezt viszont ellensúlyozza a nagyobb ellenállóképesség a β-laktamáz enzimekkel szemben.
7β-helyzet: A félszintetikus penicillineknél is alkalmazott amino-, hidroxi- vagy karboxi-benzil származékok a kefalosporinoknál is megtalálhatók, a koraiaknál a megfelelő tienil származék is. A 80-as évek óta azonban szinte egyeduralkodó az (2-aminotiazolil)-2-metoximinoacetil oldallánc a 3. és 4. generációs kefalosporinokban. Kis módosításoktól eltekintve, ennél a már lassan 25 éve használt oldalláncnál jobbat azóta sem találtak.

Hatástani szempontból az egyértelmű csoportosítás szinte lehetetlen. Megkülönböztethetünk metabolikusan stabil vagy nem stabil, β-laktamázzal szemben ellenálló vagy sem, orális vagy parenterális kefalosporinokat, s végül de nem utolsósorban az antibakteriális spektrum szerint 1-5 generációsokat. Egy általánosan elfogadott csoportosítást mutat be a fenti ábra, amelybe a legfontosabb származékokat foglaltuk bele.
Az első generációs kategóriába az idejétmúlta, a 60-as évek közepén bevezetett és ma már nemigen használt kefalosporinok tartoznak. Ezek erősen hatnak Gram-pozitív baktériumokra és közepes-jól a β-laktamáz enzimet nem termelő Gram-negatívakra. A 3-as helyzetben O-acetil csoportot tartalmazók in vivo nem stabilak, mivel az emlős szervezetekben jelenlevő aspecifikus észteráz enzimek a majdnem hatástalan 3-hidroxi származékká alakítják. Néhány képviselőjüket a következő ábra mutatja. Ma már nemigen használják ezeket.

A második generációs kefalosporinok szerkezetileg többfélék lehetnek, antibakteriális spektrumok is különböző. E csoport tagjai között már jó H. influenzae, B. fragilis, Legionella ellenes hatású vegyületek is vannak, az E. coli elleni hatás változó, és nem hatnak jól Ps. aeruginosa ellen. A cefuroxime volt az első metoximino csoportot tartalmazó kefalosporin, ami a kefem váznak jelentős ellenállást biztosított a β-laktamáz enzimekkel szemben:

A harmadik generációs kefalosporinoknál jelenik meg a (2-amino-5-tiazolil)-alkoximinoacetil oldallánc, amely, mint már említettük, jelenleg is a legáltalánosabban használt. Tulajdonképpen a cefotiámnál már alkalmazott 2-aminotiazol és a nokardicinekre jellemző metoximino csoport empírikus összeházasításából alkották. A metoximino csoport syn és anti konfigurációjú lehet, a megfelelő kefalosporinok bioaktivitásának aránya kb. 1:10 a syn izomer javára. A harmadik generációs cefalosporinok igen jó Gram-negatív baktérium ellenes hatással rendelkeznek, viszont változó a staphylococcusok és egyes anaerob törzsek elleni hatás. Egyes származékok, pl. a cefoperazon és cefsulodin kifejezetten hatásos Ps. aeruginosa ellen.

Az oldalláncnak megfelelő acilező karbonsav előállításának kulcslépése egy halogénketon reakciója tiokarbamiddal, ami régi rutinreakció a tiazolok előállítására, ez esetben viszont a konkrét reakciókörülmények lényegesek, mivel ezáltal befolyásolható a keletkező két izomer aránya:
-alkoximinoacetil oldallánc kialakítása")
Az általában tritil vagy klór-acetil csoporttal védett tiazol származékot vagy a peptidkémiából is ismert aktív észter módszerrel (pl. DCCI + 1-hidroxi-benztriazol), vagy miként az ábrán is látható, igen gyakran in situ készített Vilsmayer reagenssel kapcsolják az előzetesen szililezett 7-ACA származékhoz. Az egyetlen újabb, hatástanilag némileg kedvezőbb variáció e vegyületeknél az egy nitrogén atommal többet tartalmazó, 5-amino-4-tiadiazolil oldallánc.
A harmadik generációs cefalosporinok igen jó Gram-negatív ellenes hatással rendelkeznek, viszont változó a Staphylococcusok és egyes anaerob törzsek elleni hatás. Egyes származékok, pl. a cefoperazon és cefsulodin kifejezetten hatásos ps. aeruginosa ellen. A cefsulodine az ún. szűkspektrumú kategóriába tartozik: igen erős hatású, de csak Pseudomonasok ellen. A cefotaxime, ceftriaxone, cefpirome és a cefepime jól hatnak rezisztens st. pneumaniae ellen is. Említésre érdemes még a csak állatgyógyászatban alkalmazott ceftiofur is: e vegyületetnek az igen szigorú FDA is zöld utat adott a szarvasmarhák stb. légúti megbetegedéseinek kezelésére, ugyanis gyakorlatilag egyáltalán nem kerül be a tejbe (< 50 ppb).
Ha az aminotiazol oldalláncú kefalosporinok 3-as helyzetébe kvaterner nitrogént tartalmazó csoportot viszünk be, a bioaktivitás igen megnő Streptococcusok és Staphyloccoccusok ellen (meticillin rezisztensek ellen is), de egyéb Enterobacter, Citrobacter stb törzsekkel szembeni is. Ezeket az igen széles sávú kefalosporinokat szokás 4. generációsoknak nevezni. A cefpirome, cefepime és ceftazidime az eddig legismertebbek, azonban jónéhány további származék van kibocsátás előtt. A cefpirome, cefepime és ceftazidime az eddig legismertebbek. Az N-metil-tetrazoliltio oldalláncot ezekben már nem alkalmazzák az időközben kiderült mellékhatások miatt. A 7-es helyzetben a 2-amino-5-tetrazol oldallánc mellett megjelenik az 5-amino-4-tiadiazolil gyűrű is (cefclidin, cefozopran, cefluprenam). A zwitterionos szerkezet jóvoltából ezek a vegyületek elég könnyen átjutnak a sejtmembrán porinjain. Ellenállóképességük a széles spektrumú ß-laktamázokkal (ESBL) változó.

Az utóbbi években egyre több gondot okoznak az ún. MRSA törzsek által okozott fertőzések (MRSA: "meticillin rezistens St. aureus", vagy használható "multirezisztens St. aureus" értelemben is). Az MRSA törzseknél kimutatható, hogy bizonyos penicillinkötő fehérjék (PBP) megváltozása miatt az antibiotikum molekula affinitása erősen lecsökkent ezek iránt. Igen erőteljesek a kémiai fejlesztések ennek legyőzésére, s ezek alkotják ötödik generációs kefalosporinokat. Az eddig használatba vettet mutatja a következő ábra. A ceftozolon a Pseudomonas törzsek ellen kiterjezstett hatással rendelkezik.

A következő ábrán láthatjuk az orálisan is jól alkalmazható származékokat. Ezek a 7-es helyzetben metoximino-aminotiazol vagy a penicillineknél is jól ismert amino-fenilacetamido oldalláncot tartalmazzák, a 3-as helyzetben metoxi, egyszerű alkil vagy alkenil csoportot, vagy klór atomot tartalmaznak. Újabb keletűek a 3-as helyzetben vinil csoportot tartalmazó származékok, ezeknél a szabad sav is jól felszídik a gasztrointesztinális traktusból. Mint látható, több egyébként csak parenterális kefalosporinból aktív észter kialakításával (cefatemat, cefpodoxime stb.) jó orálisan is alkalmazható származékot lehet kialakítani. A per os szedett cefcapene igen jól behatol a bőrbe is, ezért bőrfertőzések esetén is alkalmazható.


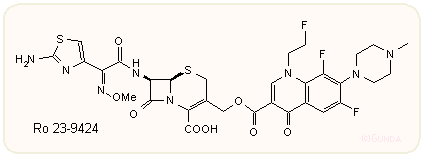
A gyógyszerkémiában azok a törekvések, amikor két külön-külön jó hatású molekulát kémiailag kovalensen összekapcsolnak a még jobb hatás reményében, 99%-ban kudarcra vannak itélve. Azonban egy ritka kivétel az először a Hoffmann-LaRoche által előállított érdekes kefalosporin kinolonkarbonsav kombinációk, például az alábbi Ro 23-9424. A kefalosporin oldalon a 3-CH2 csoport, a kinolon oldalon vagy a karbonsav, vagy az aromás N-szubsztituens (ez esetben piperazin) a csatlakozási pont, mint a példában látható cefotaxime fleroxacine kombinációban. A Gram-pozitív hatás a cefotaximra, a Gram-negatív a fleroxacinra emlékeztet. A molekula érintetlenül hatol át a a külső membránon s mint kefalosporin hat először. A hatás során, vagy a β-laktamázok hatására felszabadul a kinolonkarbonsav molekula, amely ezután a sejt belsejébe hatolva fejti ki a rá jellemző DNS giráz gátló hatást. Bár a vegyület eljutott a végső humán tesztfázisba, végül is nem lett gyógyszer belőle.