Általános szintézismódszerek
Ebben a részben konkrétan a β-laktám gyűrű szintézisének és néhány gyakran alkalmazott kulcsvegyületnek a reakcióit foglaljuk röviden össze. A β-laktám alapváz, vagyis az azetidinon gyűrű és szubsztituált származékai a β-laktám vázas antibiotikumok felfedezéséig inkább csak kuriózum volt, bár több módon is elő tudták állítani a legismertebb ezek közül Staudinger 1907-es módszere. E téren azóta óriási számú publikáció született, amit itt lehetetlen akár csak részben is összefoglalni: a közismert Houben-Weyl féle monográfia sorozat 1991-ben kiadott 16B kötete kereken 900 oldalon át tárgyalja a β-laktám gyűrű kialakítását és közvetlen reakcióit.
A β-laktám gyűrű kialakítását, bezárását négy módon tehetjük meg, ha a záráshoz csak egy kötés kialakítása szükséges, ehhez jön még az a két lehetőség, amikor egyszerre két szembeni kötést alakítunk ki (cikloaddíció).

Az N-1 C-2 kötés zárásának egyik gyakori módszere a diciklohexil- (vagy egyéb) karbodiimides módszer, a penicillin első, Sheehen féle totálszintézisét is ezzel oldották meg. Hozama változó, eléggé függ a konkrét molekulától. A karboxi csoportot vegyes anhidrid képzéssel is lehet aktiválni (mezil-, trifluoracetil, foszforhalogenidek stb). Újabban terjedt el a Mukaiyama reagens (bisz-(2-piridil)-diszulfid + trifenilfoszfin) használata. A peptidkémiában használt egyéb reagensek is sokszor sikerrel alkalmazhatók. A Grignard reagens vagy izobutil-aluminium az észterek bezárására alkalmasak
Ellentétben az előző módszerrel, a C-2 – C-3 közti kapcsolat létrehozása sokkal nehezebb és alig van rá gyakorlati példa. Ezzel szemben a C-4 – N-1 zárás ismét sokat használt gyakorlati módszer. Főleg halogén-, mezil- vagy hidroxi-származékokat lehet sikeresen alkalmazni, az utóbbiakat Mitsunobu körülmények között. A Mitsunobu reakció főleg akkor sikeres, ha a nitrogén atomhoz olyan szubsztituens csatlakozik, amelyik az amid hidrogént kissé savassá teszi.
A C-3 – C-4 kötés zárása megint ritkábban alkalmazott módszer. Általában karbén beépüléssel (diazovegyületekből), oxidatív vagy gyökös módon (CAN, HSnBu3 stb), vagy nukleofil szubsztitúcióval hajtják végre. Az utóbbi többnyire akkor lehetséges, ha a C-4 atomhoz erősen elektronszívó csoportok kapcsolódnak.

A legtöbbet tanulmányozott egylépéses β-laktám kialakítási módszerek talán a különböző [2+2] cikloaddíciók. E reakcióknak csak egyrésze valódi többcentrumú cikloaddíció, másrésze csak formálisan az, vagyis a két kötés igazából nem egyszerre alakul ki és lehetséges egy ionos köztitermék fellépése. A pontos mechanizmust csak gondos vizsgálatokkal lehet tisztázni. Mivel többnyire két új aszimmetriacentrum keletkezik, négy termék keletkezése várható, melyeket az R, S nomenklatúrán túlmenően általában röviden csak a cisz/transz α/β megjelöléssel azonosítanak. Ezek aránya nagyon függ a szubsztitúciós viszonyoktól és a reakció körülményeitől, és a kívánt sztereokémiai végeredmény miatt gyakran alkalmaznak királis szubsztituenseket. Mint a korábbi fejezetekben láthattuk, a 3-amino-azetidinonoknál cisz (penicillin, kefalosporin, monobaktám típus), a 3-alkil azetidinonoknál transz a kívánt konfiguráció (karbapenem típus).
A két lehetőség közül az első, amikor egy megfelelő olefint egy izocianáttal reagáltatják. Klórszulfonil- vagy triklóracetil-izocianát a legkedveltebb reagens, a cikloaddíció után a klórszulfonil csoportot reduktív úton (Na2so3, Zn + NH4Cl stb), a triklóracetil csoportot átacetilezéssel lehet eltávolítani.
Staudinger reakció (ketének cikloaddíciója)
A Schiff-bázisokból, iminekből több módon is kialakítható a β-laktám gyűrű. A keténekkel, illetve savkloridokkal vagy egyéb aktivált savszármazékokkal bázisok jelenlétében történő [2+2] cikloaddíció Staudinger reakció néven régen ismert folyamat. A penicillin kémiát szemelőtt tartva elsőnek Bose és munkatársai tanulmányozták igen behatóan az azido-acetilklorid és iminek reakcióját; ezt a variánst egyébként Bose-reakciónak is nevezik.

Az ábrán látható, hogy a ketén komponens kiindulási anyaga, amennyiben 3-amino-azetidinon származék a végcél, azido-acetilklorid, ftaloil-glicilklorid, egy β-dikarbonil vegyülettel (pl. acet-ecetsav etil-észter) védett glicin származék (Dane-só) vagy más hasonló lehet.. A képződött 3-azido-azetidinont enyhe redukálószerrel, pl. SH2 lehet aminná alakítani, a ftalimido csoport bontására a metil-hidrazin a legalkalmasabb. A Dane származékból az amin enyhe savas hidrolízissel szabadítható fel. Újabban kedvelt még a negyedik vegyület, egy optikailag aktív (4S)-fenil-oxazodilil-ecetsav származék (ill. az ebből kapható ún. Evans-Sjögren ketén), ennek viszont az eltávolítása körülményesebb (Li + NH3). A karbapenemekre jellemző C-3 oldalláncot tartalmazó vegyületekhez alkilkarbonsavak és hidroxi-alkilkarbonsavak kellenek, pl. az ábrán utolsó 3-(tri-izopropil-szililoxi)-vajsav. A savkloridok természetesen nem állíthatók elő mindig, ilyenkor az aktiválásra más savszármazék is szóba jöhet, például vegyes anhidridek, foszfor-reagensek (PPh3 + CBr4, (PhOP(O)Cl2, (EtO)2P(O)Cl,), trifluor-ecetsav anhidrid, karbonil-diimidazol, SOCl2 + DMF stb.
A reakció sztereokémiáját sok faktor befolyásolja és ezért nehéz előre megjósolni az eredményt. A sztereospecifitást királis kiindulási anyagokkal, segédcsoportokkal lehet elősegíteni. Ezek három helyen alkalmazhatók: a kiindulási savban, az imin C-atomján (aldehid komponens) és az imin N-atomján (amin komponens). A hatás feltérképezésére igen sok származékot vizsgáltak. Általánosságban elmondható, hogy a sav vagy aldehid komponens kiralitásának nagyobb befolyása van a sztereospecifitásra, mint az amin komponensnek. Királis aldehidként a könnyen előállítható D-glicerinaldehid vagy hasonló cukorszármazékok kedveltek. Királis aminként aminosav származékok, fenil-etilamin stb jöhetnek szóba. Természetesen több királis prekurzor kombinálható is. Schiff-bázis helyett imidátok vagy tioimidátok is használhatók, ez inkább a transz izomerek keletkezésének kedvez, főleg ciklikus imidátok esetén.
Ma már általánosan elfogadott, hogy egy keténnek illetve savkloridnak és egy iminnek a reakciója kétlépcsős, először egy zwitterionos köztitermék (a) jön létre, és nem szinkron többcentrumú cikloaddíció játszódik le. A köztiterméket sikerült spektroszkópiai úton kimutatni vagy megfelelő reagenssel elfogni:

A ketén támadása ennek térbelileg kevésbé zsúfolt oldala felől (H atom vagy a kisebb szubsztituens) indul olymódon, hogy a karbonil oxigén atomjának LUMO-ja (amely koplanáris az R1 szubsztituenssel) ortogonálisan közelít az imin felé. A létrejövő zwitterionos köztitermék [2+2] konrotációs gyűrűzárással ciklizálódik úgy, hogy a termodinamikailag kedvezőtlenebb termék keletkezik, a két hidrogén vagy kisebb szubsztituens egymáshoz képest cisz helyzetbe kerül. Ha a ketén "fordítva" közeledik az iminhez, a másik cisz izomer képződik. Az aciklikus iminek természetesen a kedvezőbb E térállásban vannak, azonban ciklikus imineknél a két szubsztituens Z térállású, ilyenkor konrotációnál a transz végtermék a kedvezményezettebb.

A zwitterionos köztitermékre több kísérleti adat, például oldószerhatás, és elméleti, ab initio és szemiempírikus kvantumkémiai számítások utalnak. A keletkező izomerek eloszlása pusztán sztérikus effektusokkal nem magyarázható kielégítően. A konrotációs gyűrűzárulás hasonló a ciklobutének elektrociklusos nyílásához/csukódásához és ab initio kvantumkémiai számítások szerint alkalmazható rá a Houk-féle ún. elektronikus forgásszelektivitás (torquoelectronic effect). Eszerint a reakció során a konrotáció forgásiránya úgy alakul, hogy egy elektronszívó csoport befelé, egy elektronküldő csoport kifelé mozduljon el. Másszóval, egy elektronszívó S csoportot tartalmazó vegyület olyan konformációt és forgásirányt igyekszik felvenni, mint az a következő ábrán látható, a kérdéses csoport az Sb helyzetnek felel meg. Ennek az effektusnak az irányító hatása erősebb lehet, mint a térbeli effektusok (kontrasztérikus szelekció). A zwitterionos "kifelé" és "befelé" átmeneti állapot stabilizálásánál jelentős lehet az oldószerhatás is. E három effektus sztérikus, elektronikus-forgató és oldószer egyenlegéből alakul ki a Staudinger reakció sztereokémiai lefutása.
 >
>A ketének in situ előállítására és iminekkel való reakciójára alkalmas még Hegedus módszere is (ld. korábbi nagy ábra), amelyben króm alkoxikarbén komplexeket imin jelenlétében fotolizálnak. Ekkor a fém-szén kötésbe szénmonoxid épül be, s a keletkezett komplex keténként viselkedik, azonban szabad ketén nincs jelen, mivel az erre utaló jelek (ketén dimerizációja, stb.) nem tapasztalhatók. Az igen diasztereoszelektív folyamat végeredményéül jó hozamban képződnek a ß-laktámok.
Enolátok és iminek reakciója
Meg kell még említeni a különböző enolátok és iminek kondenzációs reakcióit is. Elsősorban lítium-, cink-, ón-, titán- és trimetil-szilil-enolátokat alkalmaznak, az amin komponens sokféle lehet, újabban kedveltek az in situ preparált trimetil-szilil-iminek, például:
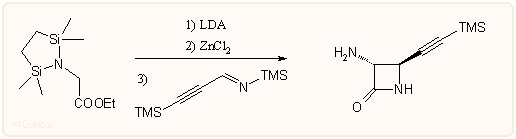
A reakció során majdnem tisztán a transz izomer keletkezik, a megfelelő lítium enolátot alkalmazva pedig a cisz izomer a kedvezményezett. A fém ion (Na, Li stb) is nagyon befolyásol. A reakciók sztereospecifitása függ attól, hogy Z vagy E enolátról van-e szó, a sztérikus kölcsönhatásoktól stb. Az energetikailag legkedvezőbb kád alakú átmeneti állapotban az R1 és R3 szubsztituensek térbeli helyzete a Z enolát és Z imin esetén a legkedvezőbb, ez a cisz terméket eredményezi (ábra). Elméleti számítások szerint is a Z elrendezésű komponensek alkotta szék állapot 7-8 kcal/móllal stabilisabb a többi lehetséges állapotnál.

Kulcsvegyületek
Végezetül két gyakori köztitermék, kulcsvegyület fontosabb reakcióit foglaljuk össze különösebb kommentár nélkül. Az első a már sokat említett diszulfid származék, a sokrétűen továbbalakítható Kamiya-diszulfid:

A másik ábra a 4-acetoxi-azetidinon reakcióit mutatja be. Előállításáról és felhasználásáról már szóltunk a karbapenemeknél. Itt kiemelnénk, hogy az acetoxi csoport nemcsak a szokásos nukleofil szubsztitúcióval cserélhető, hanem fémorganikus vegyületekkel, ón és szilil enolátokkal, triflátokkal, stb. változatos sztereospecifikus reakciókra késztethető. Jelentőségét mutatja, hogy a szerves vegyipar nagy mennyiségben is előállítja, főleg a karbapenem szintézisekhez használható védett 3-hidroxietil származékot.
